
Theoretical & Computational study of Polymer and Nanomaterial Lab.
Significant advances in the field of computer simulations (both methodological and algorithmic) and the rapid increase in computing power have led to the development of powerful multi-scale simulation tools (i.e., from the microscopic quantum/atomistic level via mesoscopic coarse-graining procedures all the way up to the macroscopic or continuum level). These developments have the potential to address fundamental issues in materials science and technology by revealing the molecular origin of unique phenomena exhibited at the macroscopic level and by elucidating the structure-property-processing relationships in systems with a complex internal microstructure of synthetic polymers and biopolymers. Motivated by these advances, our research interests are below as shown.

Atomistic configurations of C128H258 linear polyethylene (PE) melt under shear flow.
[Weissenberg number (product of the longest relaxation time of system and imposed shear rate) Wi = 100]
Research topic 1: Bulk polymer viscoelasticity
-
Despite numerous experimental and theoretical efforts to elucidate the role of chain architecture (i.e., chain-branching) on polymer rheology, a sound microscopic-level understanding is still missing. We will address this issue through direct atomistic Nonequilibrium molecular dynamics (NEMD) simulations both in shear and elongational flows, which will help us collect detailed information about local structure and dynamics at various length and time scales. Various kinds of polymers will be studied: (a) long-branched polymers with different degrees of branching, i.e., H-polymer, (b) short chain branched (SCB) polymers with different branching frequency and branch length, and (c) ring polymers of varying size. The last type of polymer has recently attracted considerable attention due to its peculiar rheological properties (e.g., absence of distinct plateau modulus) and our low level of understanding of the large-scale relaxation mechanism (e.g., no chain reptation). Both transient and steady-state properties will be investigated, and a number of rheological models and proposed mechanisms will be critically tested.


(Left) Schematic illustrations for the fundamental molecular mechanisms of an individual chain for linear,
H-shaped (H), and short-chain branched (SCB) polymer systems. (Right) Steady state viscosity as a function
of reduced shear rate for linear (C178H358), H (C178H358), and SCB (C178H358) PE melt systems.
-
NEMD simulations have been widely applied to dense polymeric systems under various flowing conditions. However, due to the lack of a proper periodic boundary condition, the current NEMD method cannot simulate uniaxial elongational flow, which together with the shear flow is one of the most important flow types for characterizing material properties and developing a general constitutive model in polymer rheology. To overcome the shortcomings of NEMD, a nonequilibrium Monte Carlo (MC) methodology was developed based on the GENERIC (General Equation for the Non-Equilibrium Reversible-Irreversible Coupling) thermodynamic formalism for simulating flowing unentangled polymer melts with reasonable computational costs. Our recent developments of MC techniques to efficiently generate a large matrix of the short-chain branched polymers, together with our NEMD and GENERIC-MC methods, will be applied to examine the global structural/dynamical behaviors of the polymeric systems with/without external flow fields.


(Left) Representative atomistic snapshots of the flow-induced crystalline phase created under, alpha_xx=1.0 at T=350 K.
(Right) Comparison of the structure of the flow-induced crystalline phase developed in the liquid at T=350 K with
the X-ray diffraction experimental data of a n-eicosane crystal.
-
Molecular simulations [molecular dynamics (MD) and Monte Carlo (MC) methods] are preferred as the optimum tool for exploring the fundamental structure–property relationships in polymer rheology because they can control a well-defined molecular architecture to suit various requirements. Therefore, many important dynamic aspects at molecular levels have been revealed by NEMD simulations for linear and branched polymer melts under equilibrium or nonequilibrium conditions. On the other hand, simulation for dilute polymer solution is hard to carry out at molecular-level simulation because huge amounts of water molecule require expensive computational costs. To reduce the expensive computational costs, the coarse-grained Brownian Dynamics (BD) simulations [bead-spring and bead-rod] are used to simulate the dilute polymer solution, which is informative for study about microscopic dynamics of single polymer chain without inter-chain interaction.
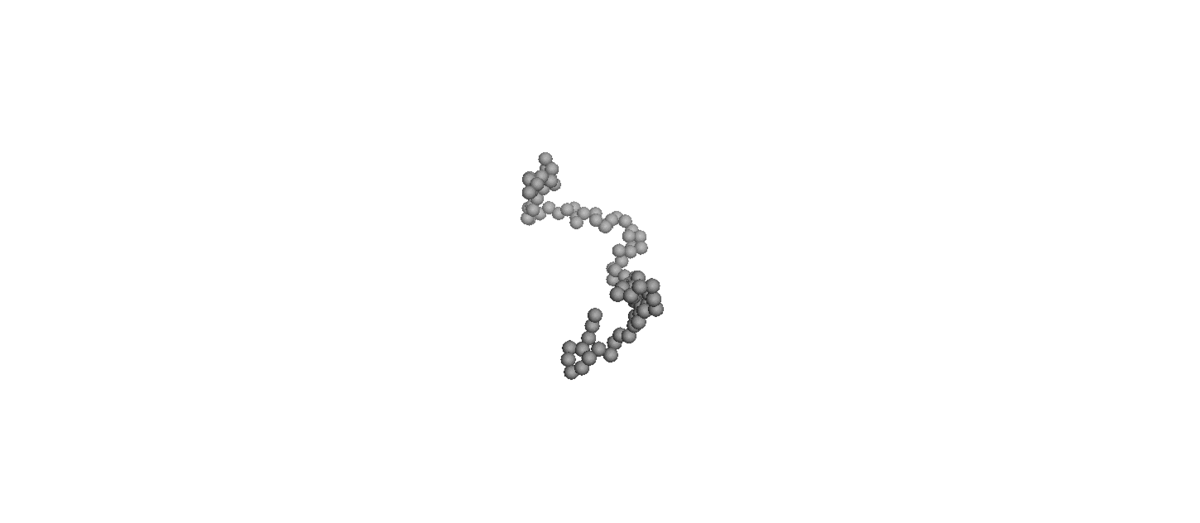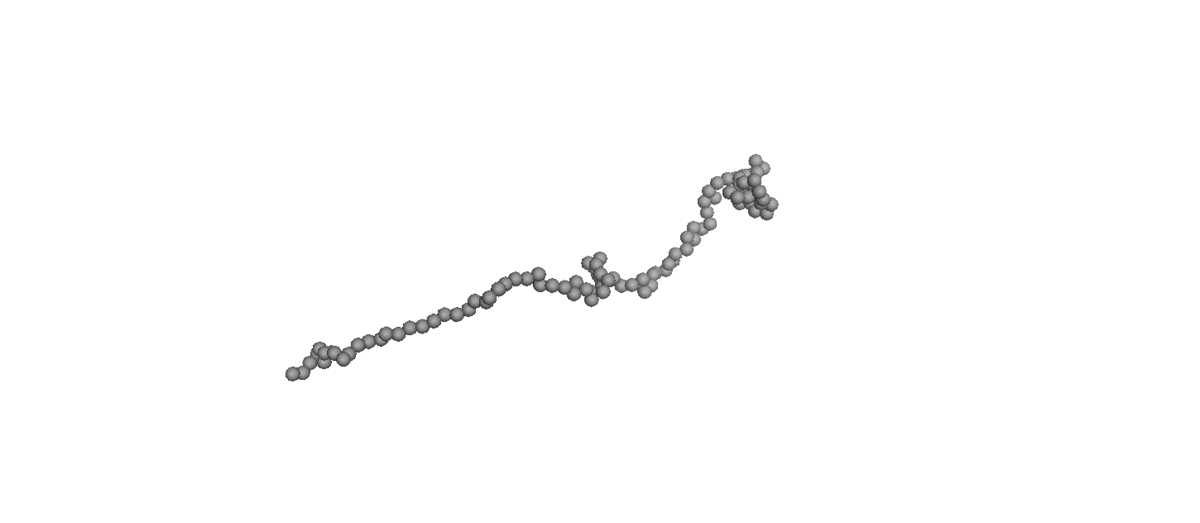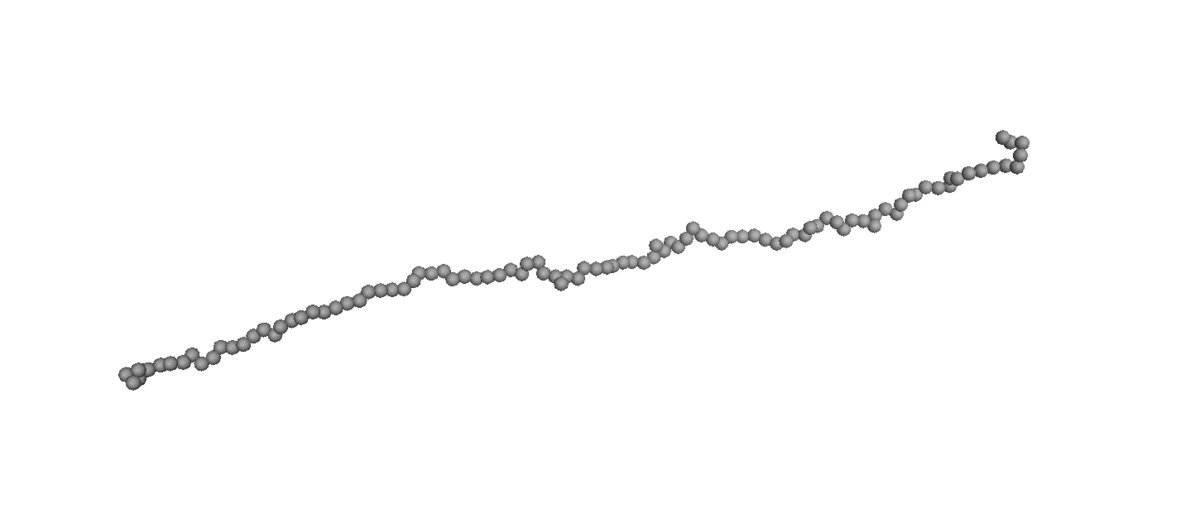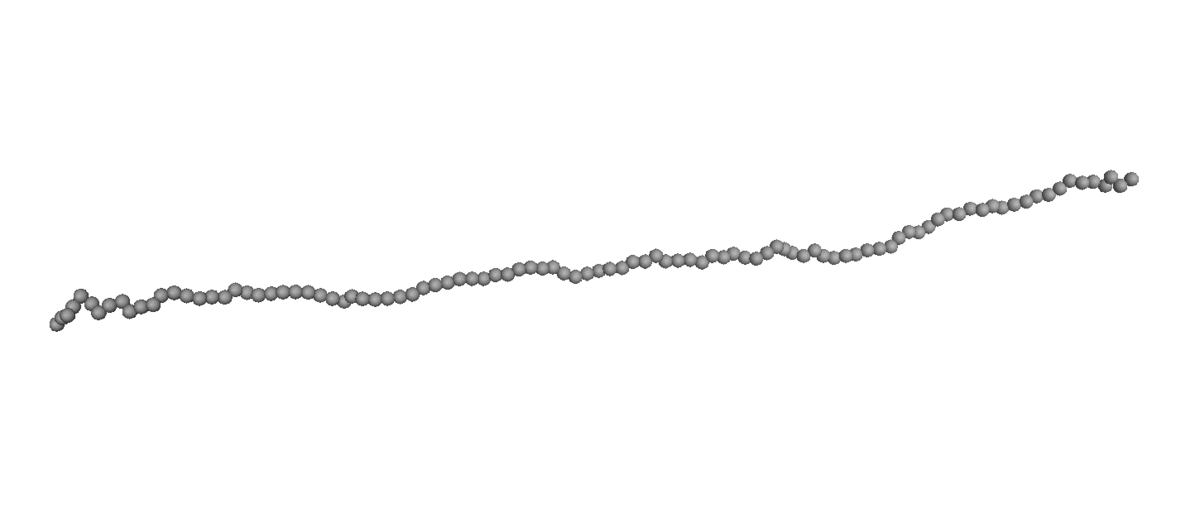


(Left) Coil-stretch dynamics of a linear polymer chain in dilute solution under shear flow (Wi = 100). (Right) Steady-state viscosity for Ring
and linear polymer systems obtained from atomistic NEMD and mesoscopic BD simulations as a function of Wi under shear flow.
-
In the long term, systematic efforts will be undertaken to extend all of our atomistic MC/MD/NEMD/GENERIC-MC simulations and coarse-grained simulations, together with theoretical modeling of various materials, which have not been yet well understood or which are not yet explored. Detailed accounts from both thermodynamic and rheological viewpoints will be taken when analyzing the simulation data and predicting and modeling the structural and dynamical evolution of systems.


(Left) Atomistic configurations of C128H256 ring PE melt under shear flow (Wi = 100). (Right) Schematic description of the characteristic
molecular mechanisms for the interfacial linear, ring, SCB linear, and SCB ring polymers in the three representative flow regimes
(weak, intermediate, and strong).
Research topic 2: Interfacial structure and rheology of polymeric fluids
-
Nonequilibrium molecular dynamics (NEMD) simulations with parallel, multiple-time step algorithms will be employed to probe the structural and rheological behaviors at the nano- and micro-scale for selected polymer-substrate systems. The simulations will help us elucidate the fundamental molecular mechanisms of polymer chains at solid surface under flow. This will help us systematically analyze the macroscopic slip phenomena and how they are affected by the polymer architecture and substrate, which provides molecular-level understanding of slip phenomena and the associated interfacial instabilities (e.g., melt fracture). We will probe the response of the system over a wide range of forcing levels, which will help us determine the transition from no-slip to slip conditions, as quantified by the relationship between slip length and wall speed. The critical shear rate for the transition from defect or localized slip to global slip will also be determined. In case slip is observed, we will quantify the conditions under which global slip takes place and the relationship between slip length and wall speed. This relation will be utilized to suggest a new type of boundary condition in theoretical modeling and numerical simulations and to compare against available expressions reported recently in the literature through, e.g., a thermodynamic description of deforming interfaces. Molecular-level understanding of the underlying dynamics mechanism of polymer molecules with/without external fields will be of great practical importance in many applications. Combination of NEMD with coarse-grained models will be necessary in this research.
Research topic 3: Nanostructured polymeric materials
-
The real systems used for experiments and daily life are too large to approach in the view point of atomistic or coarse-grained schemes, so continuum-level simulations need to imitate the structural and rheological phenomena of real systems (i.e., water-drop experiment, pressure sensor). Continuum-level simulations are carried out by commercial software (i.e., COMSOL Multiphysics, ABAQUS) with a variety of simulation modules. Based on thermodynamic and rheological viewpoints obtained from atomistic and coarse-grained simulation, continuum-level simulation can be of great help in many applications, such as development of new polymeric materials with intended structural and rheological properties. It is quite difficult to carry out overall procedures to produce a versatile sensor covering from the research on specific materials in details and controlling their micro-structures through only experiments, since there are countless number of cases to consider as candidates and many infeasible measurements throughout the micro- to nano-scale (e.g., the contact area, stress propagation and concentration, electric field distribution, and etc.). Multi-scale simulations collaborated with experimental approaches can model and fabricate nanostructured polymer composite materials with highly improved abilities. Through a precise modeling with multi-scale simulations collaborated with experimental processing, the most optimized construction of the nanostructured polymer composite materials with highly improved sensing performances (e.g., ultrahigh sensitivity, linear sensing capability, broad sensing range, etc.) would be possible.
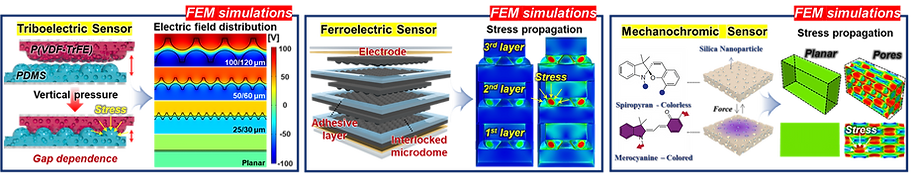
Continuum-level simulation of polymer nanostructured sensors using finite element method (FEM).
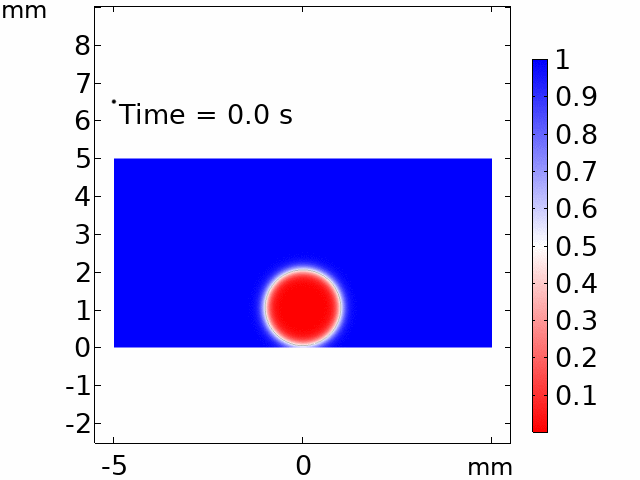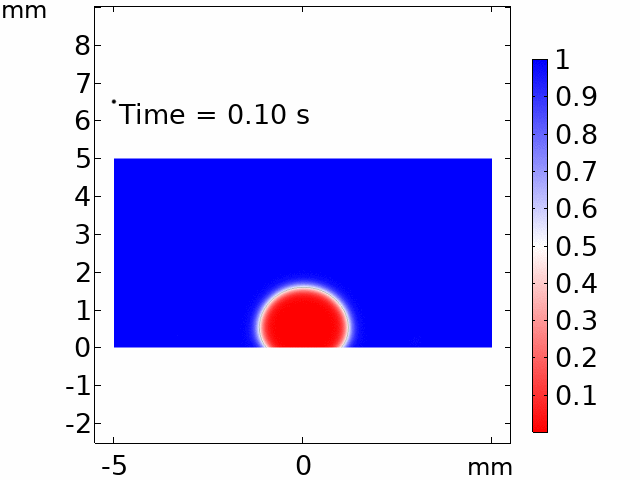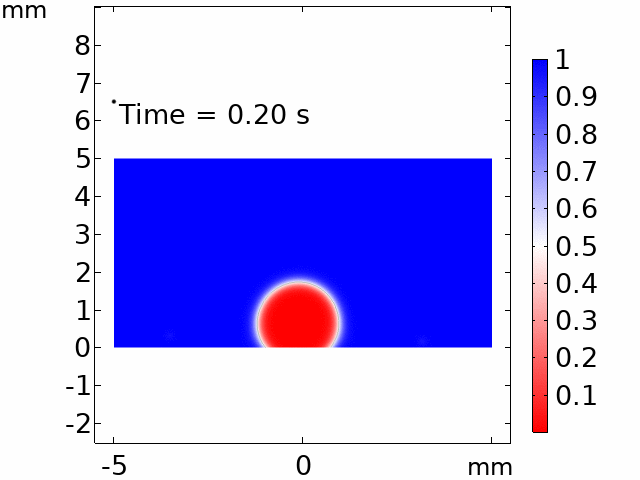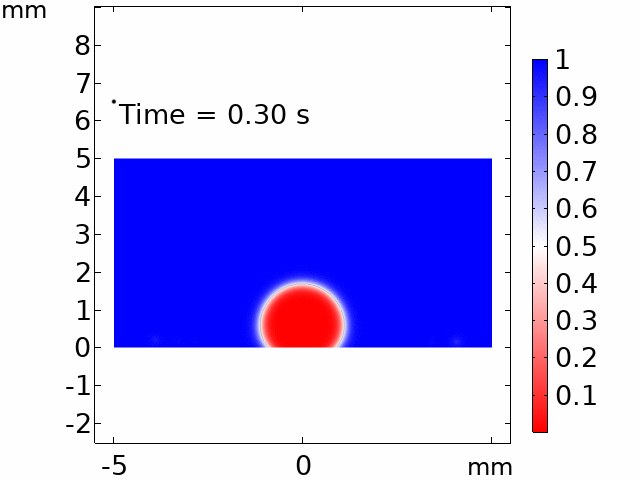
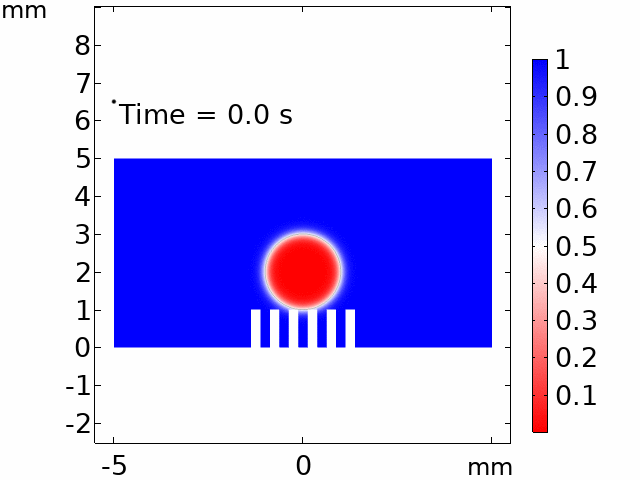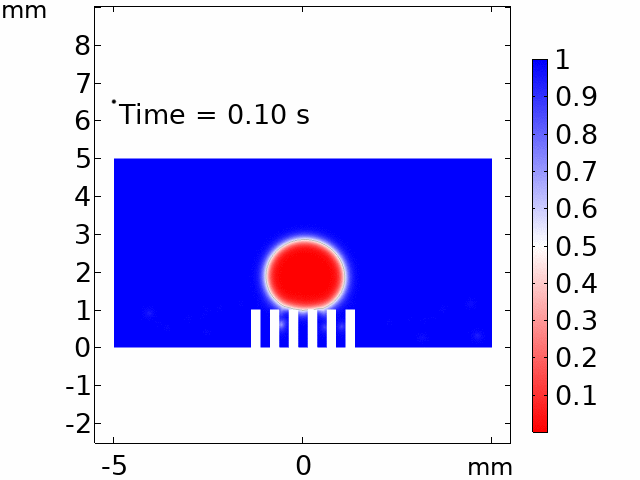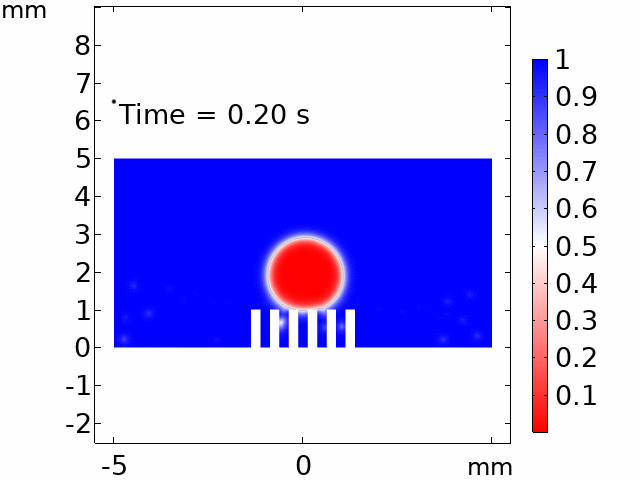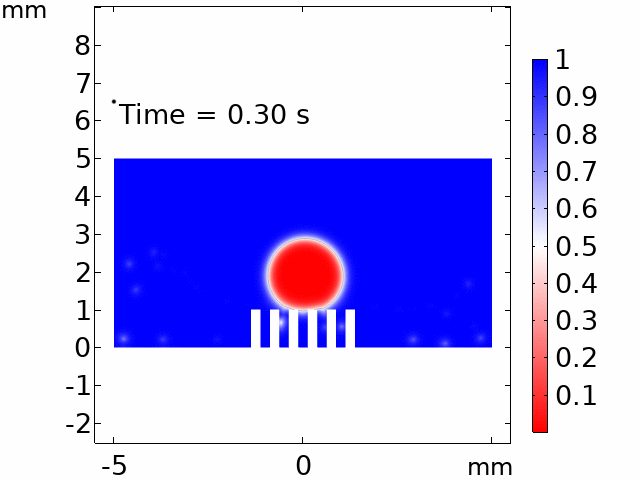
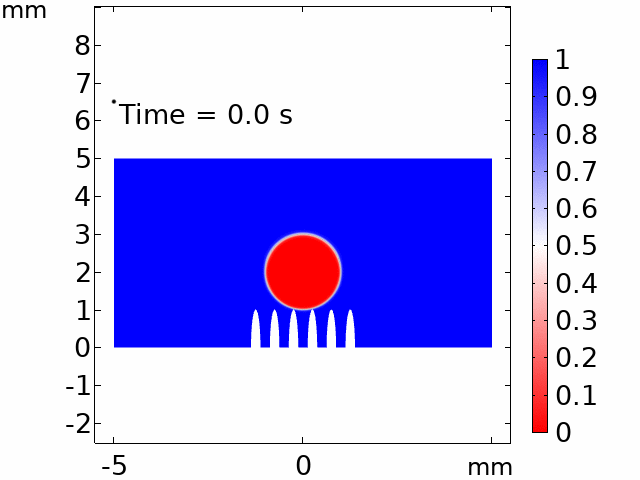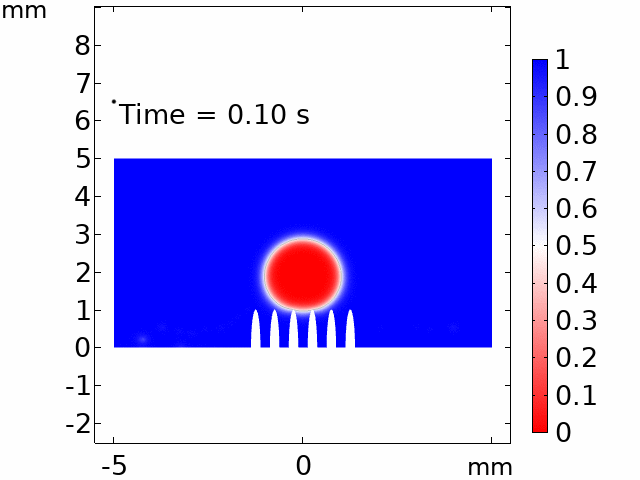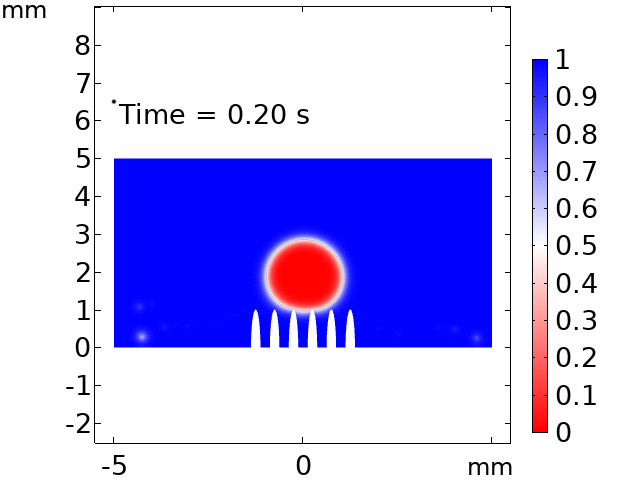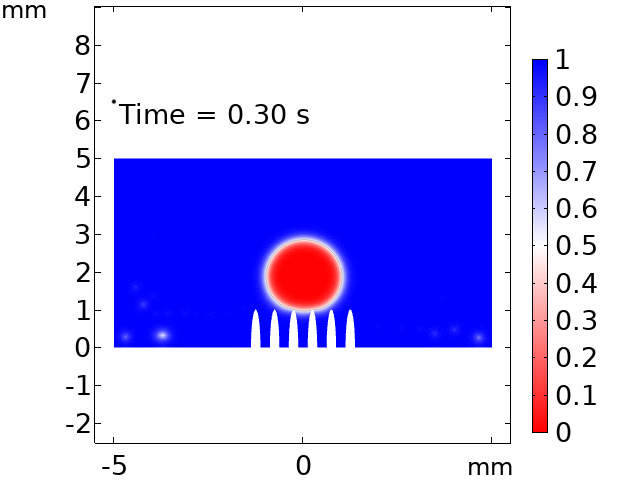
Macroscopic simulations of water-drop on various nanostructured surfaces via COMSOL Multiphysics.